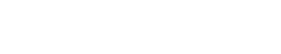






Juan tiene 23 años y padece síndrome de Down (SD). Acude a la consulta acompañado por su madre para solicitar una analítica de control con hormonas tiroideas. Hace 5 años fue diagnosticado de hipotiroidismo y sigue tratamiento con 75 mg de levotiroxina. Por lo demás, está relativamente sano, visita poco la consulta y su discapacidad intelectual no le impide acudir a trabajar a un taller ocupacional. En su historia clínica apenas se menciona que presenta un discreto sobrepeso y que utiliza gafas desde hace años. Su madre interviene para indicar que Juan se las quita con frecuencia y que cree que ha perdido visión. Se objetiva una agudeza visual corregida de 0,4 bilateral y que sólo mejora hasta 0,5 con el estenopeico. El fulgor pupilar parece disminuido en ambos ojos. Se decide pedir la analítica habitual, remitir al oftalmólogo y citarle de nuevo para revisar los resultados y realizar una serie de actividades preventivas.
Puntos clave
- El SD es la causa genética más frecuente de retraso mental.
- El factor de riesgo fundamental es la edad materna avanzada en el momento del embarazo.
- El número de recién nacidos con SD disminuye de forma constante como consecuencia de los programas de cribado, aunque el número de afectados se mantiene debido a la mayor supervivencia.
- El cribado en el primer trimestre consigue detectar a más del 90% de los fetos con SD.
- El diagnóstico prenatal se realiza por medio del cariotipo de las células fetales obtenidas a través de una biopsia de vellosidades coriales o de una amniocentesis.
- La realización de estas técnicas diagnósticas exige asumir un 1% de pérdidas fetales.
- La mayoría de las personas con SD sufre un retraso mental leve o moderado.
- Existen patologías muy prevalentes en el SD y que en ocasiones entrañan un diagnóstico dificultoso. Diferentes guías aconsejan su cribado con grados de recomendación B-C.
- Estas patologías prevalentes suelen manifestarse en forma de trastornos conductuales. Su presencia obliga a mantener un alto grado de sospecha para facilitar el diagnóstico.
Introducción
El médico inglés John Langdon Down publicó, en 1886, un artículo que describía minuciosamente a un grupo de pacientes con discapacidad intelectual y características físicas parecidas1. En 1958, el genetista francés Jérôme Lejaune relacionó el síndrome de Down (SD) con una modificación cromosómica en el par 21.
Se pueden dar tres tipos de alteraciones cromosómicas que expresen el fenotipo característico del SD: trisomía 21 (94% de los pacientes), en la que todas las células portan un cromosoma 21 de más; translocación (4% de casos), en la que el material genético del cromosoma 21 adicional se adhiere a otro cromosoma, y mosaicismo (2% de casos) en el que solamente algunas células poseen el material cromosómico excedentario.
Epidemiología
El SD es la causa genética más común de retraso mental, supone una cifra cercana a 1 por cada 1.000 nacidos vivos, y su incidencia depende de dos circunstancias divergentes:
- Por una parte, es conocido que el riesgo de tener un niño con SD aumenta de forma gradual con la edad materna, siendo el incremento exponencial a partir de los 35 años (figura 1)2. Así el embarazo tardío, cada vez más frecuente en los países desarrollados, contribuiría a aumentar el número de embriones con SD.
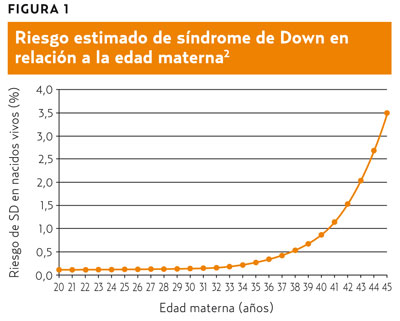
- Sin embargo, en países como España en los que se ha generalizado el acceso al diagnóstico prenatal, las interrupciones voluntarias del embarazo compensan este efecto y el número de recién nacidos con SD disminuye de forma constante (figura 2)3.Por ese mismo motivo, el número de diagnósticos prenatales aumentó un 71% en Inglaterra y Gales en el período 2007-2008 en relación al comprendido entre 1989-1990, mientras que el de neonatos con SD disminuyó un 1% en los mismos intervalos4.
En los últimos 50 años la supervivencia durante el primer año de vida ha aumentado de menos del 50% hasta más del 90%5, y la esperanza de vida supera ya los 60 años6. Estos logros compensan, en parte, el descenso de nuevos casos. Cada cupo de 1.600 pacientes contaría, de media, con un SD mayor de 14 años.
Diagnóstico
El diagnóstico prenatal del SD exige demostrar la trisomía del par 21 en las células fetales. En el momento actual, estas células sólo se pueden obtener a través de las denominadas técnicas de diagnóstico de certeza, como la amniocentesis o la biopsia de vellosidades coriales. Desgraciadamente, de ellas se deriva la pérdida fetal en cerca del 1% de las ocasiones7. Por ello, históricamente se ha pretendido limitarlas a poblaciones de riesgo, en concreto a las embarazadas añosas. Sin embargo, la aparición de una serie de marcadores bioquímicos y ecográficos permite seleccionar actualmente a las embarazadas susceptibles de ser sometidas a tales técnicas de una forma más satisfactoria.
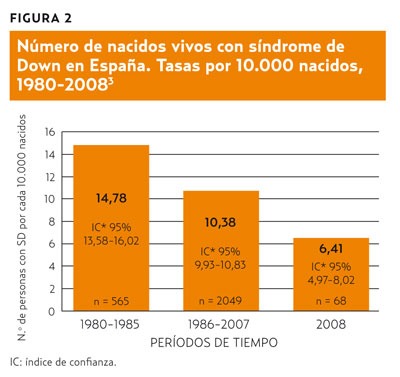
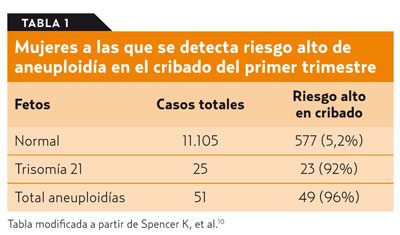
Los programas de cribado del SD pretenden ofertar a los padres la información prenatal precozmente para que puedan tomar las decisiones que consideren pertinentes. El denominado test combinado bioquímico-ecográfico del primer trimestre valora conjuntamente marcadores bioquímicos (subunidad blibre de la gonadotropina coriónica humana: fb-hCG y proteína A del plasma sanguíneo asociada al embarazo [PAPP-A]) y ecográficos (trans-lucidez nucal) junto a la edad materna para proporcionar una valoración cuantitativa del riesgo de SD que permite clasificar a los embarazos como de riesgo alto o bajo. Varios estudios han demostrado que esta prueba consigue detectar hasta un 90% de los fetos afectados8,9. En su debe hay que asumir, aproximadamente, un 5 % de falsos positivos, esto es, de embarazadas que se someterán a los riesgos de una técnica diagnóstica de certeza sin necesidad. Los resultados concretos obtenidos por Spencer et al. en la clínica OSCAR de Londres se relacionan en la tabla 110, donde se comprueba que a 577 mujeres (5,2% de las participantes) etiquetadas como de alto riesgo en el cribado se les ofertó la realización de pruebas diagnósticas invasivas a pesar de que gestaban un feto aparentemente sano.
Varios programas autonómicos contemplan la captación entre las semanas 11 y 14 de la gestación. De retrasarse la inclusión en el programa, aún puede ofertarse un cribado bioquímico durante el segundo trimestre, con una tasa de detección cercana al 65% y una tasa de falsos positivos del 5%.
El diagnóstico posnatal no suele ofrecer dificultades y al médico de familia con responsabilidades en el seguimiento pediátrico le suele llegar resuelto desde las unidades de neonatología. Existen una serie de características fenotípicas, conocidas como criterios de Hall (tabla 2)11, que aparecen comúnmente en los recién nacidos con SD. En todos los casos se pueden reconocer al menos 4 de estos rasgos y el 89% de los pacientes acumulan 6 o más. El diagnóstico ha de ser confirmado siempre a través del estudio del cariotipo.
Desarrollo psicomotor
Los niños con SD se desarrollan de modo similar a como lo hacen el resto, aunque de forma más lenta. En la tabla 312 se muestran las diferencias en los tiempos de adquisición de distintos hitos del desarrollo en relación a la población general. Los déficit no se manifiestan de un modo uniforme, sino que predominan en la denominada inteligencia analítica, que engloba el lenguaje, el manejo de símbolos o la resolución de problemas, mientras que el área social está relativamente preservada. En el área motora destacan la hipotonía generalizada y la torpeza en los movimientos que mejora parcialmente en la edad adulta.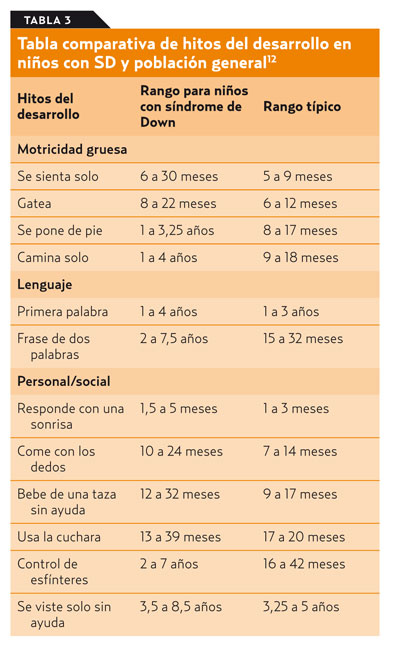
Existe consenso en admitir que todos los bebés con SD deben ser remitidos a un programa de atención temprana. Los estudios que sustentan este proceder son casos-controles con muestras de tamaño reducido (grado de recomendación B),y se basan en el concepto de neuroplasticidad, esto es, la capacidad del sistema nervioso de «madurar» a través de la estimulación y la experiencia, objetivo que se consigue en mayor medida cuanto más precoz sea la intervención. Por otra parte, la participación activa de la familia redunda en una mejor adaptación y en una disminución en sus niveles de ansiedad13.
El 97% de las personas con SD muestra algún grado de retraso mental, que en más de la mitad de los casos puede clasificarse como de grado leve o moderado. La integración escolar durante la infancia y adolescencia y la incorporación laboral persiguen alcanzar el disfrute de una vida adulta lo más libre y autónoma posible.
Patologías relacionadas
Existen una serie de patologías que ocurren más frecuentemente en los pacientes afectados de SD. El médico de familia debe conocerlas y mantener un elevado grado de sospecha para facilitar su diagnóstico, ya que las dificultades para expresarse tienden a enmascarar la sintomatología, que se manifiesta con frecuencia en forma de trastornos conductuales.
Desarrollo físico
Es característico del SD un retraso del crecimiento que conduce a una baja talla en la vida adulta. La utilización de curvas de crecimiento diseñadas para la población general no resulta adecuada, por lo que se han calculado tablas específicas. Desde el año 1998 contamos con tablas españolas que se actualizaron en el 200414. La monitorización de las alteraciones pondoestaturales durante la infancia permite sospechar la presencia de diversas patologías, ya que constituyen marcadores sensibles de la irrupción de las mismas. El sobrepeso y la obesidad son frecuentes entre los adolescentes y adultos con SD, y debieran ser controlados mediante el seguimiento de una dieta equilibrada y la realización de ejercicio físico adaptado a las condiciones de cada paciente concreto.
Hipotiroidismo y celiaquía
El hipotiroidismo y la enfermedad celíaca son dos patologías en las que coexisten la presentación asintomática o con síntomas difícilmente distinguibles de los habituales en el SD, y una frecuencia excepcionalmente alta en comparación con la de la población general. También existen procedimientos diagnósticos accesibles por lo que la mayoría de guías clínicas aconsejan su cribado periódico en los niños y adultos con SD. En el caso del hipotiroidismo, las analíticas deben mantenerse anualmente durante la edad adulta ya que los nuevos casos aparecen a medida que avanzan los años, hasta alcanzar una prevalencia cercana al 30%15. Al igual que en la población general, el manejo del hipotiroidismo subclínico es un tema controvertido. En cuanto al cribado de la enfermedad celíaca silente, el reciente protocolo de detección precoz, auspiciado por el Ministerio de Sanidad y Consumo en 2008, aconseja incluir al SD entre los grupos de riesgo basándose en la prevalencia superior al 12%. Del mismo modo, el grupo PAPPS (Programa de Actividades Preventivas de Promoción de la Salud) de la Infancia y Adolescencia recomienda el cribado repitiendo las analíticas cada 3 años. Sin embargo, Swigonski et al. publicaron en 2006 un estudio de coste efectividad en el que desaconsejaban el cribado en los pacientes con SD16. El elevado coste de cada diagnóstico de linfoma (principal beneficio del diagnóstico de la celiaquía silente), dependía de su escasa frecuencia, de la elevada edad a la que se manifiesta habitualmente y de la menor esperanza de vida de estos pacientes en relación con la población general. En cualquiera de los casos, el médico de familia ha de estar alerta para diagnosticar diligentemente los casos de enfermedad celíaca sintomática que se presenten.
Cardiopatías
La patología cardíaca congénita afecta a cerca de la mitad de los neonatos con SD. El 40% de ellos padece defectos completos del septo auriculoventricular y diferentes alteraciones estructurales que conviene diagnosticar y corregir quirúrgicamente en los primeros meses de vida, ya que la hipertensión pulmonar irreversible se desarrolla más rápidamente que en otros niños cardiópatas. Por lo tanto, los niños con SD han de ser estudiados mediante ecocardiografía en el primer mes de vida (grado de recomendación B). A pesar de ello, el 17% de una muestra holandesa de adultos con SD procedentes de instituciones residenciales padecía una cardiopatía congénita que permanecía sin diagnosticar17, por lo que parece razonable indicar una ecografía si no se había practicado anteriormente.
A partir de la adolescencia, los pacientes con SD desarrollan con gran frecuencia prolapso de la válvula mitral (hasta un 57%) y regurgitación aórtica ligera o moderada18. Debido al carácter benigno y habitualmente asintomático de estas alteraciones valvulares adquiridas, la auscultación anual cuidadosa pudiera ser suficiente para detectar los signos característicos: clic y soplo sistólico del prolapso mitral y suave soplo diastólico precoz de la insuficiencia aórtica. La principal consecuencia para este grupo de pacientes sería su inclusión, junto con los diagnosticados de cardiopatías congénitas, en la indicación de profilaxis de la endocarditis bacteriana. Podría discutirse si tal razón justificaría por sí misma la búsqueda activa mediante ecocardiografía entre los adultos asintomáticos, como proponen algunas guías clínicas (grado de recomendación C).
Alteraciones ORL y odontológicas
La prevalencia de hipoacusia entre los niños con SD es alta, siendo en la mayoría de los casos de tipo conductiva, a lo que contribuyen, entre otras, la elevada frecuencia de otitis media serosa, las estrecheces del conducto auditivo externo o la impactación de tapones de cerumen. Esto implica la necesidad de realizar cribados precoces ya que la hipoacusia compromete aún más la adquisición del lenguaje y la comunicación de estos pacientes. Es recomendable practicar pruebas auditivas cada 2 años comenzando a los 6 meses de edad. La alta tasa de recurrencia de la otitis media y la escasa eficacia del tratamiento médico aconsejan un seguimiento estrecho y un tratamiento agresivo, que puede incluir la opción quirúrgica19.
El síndrome de apnea/hipopnea obstructiva del sueño es común (30-60%) entre niños con SD20. Determinadas peculiaridades anatómicas como la hipoplasia mediofacial y mandibular, la glosoptosis, la macroglosia, la estrechez de la vía aérea superior y la hipertrofia amigdalar y adenoidea son características que, añadidas a la hipotonía generalizada, justifican esta elevada prevalencia. A medida que se van cumpliendo años, la obesidad y el hipotiroidismo pueden sumarse a todo este conjunto de factores favorecedores. Por lo tanto, es lógico suponer que la frecuencia puede incluso aumentar durante la edad adulta. La presencia de irritabilidad, estado de ánimo bajo o trastornos del comportamiento orienta al médico de familia
a tomar en consideración tal posibilidad diagnóstica. Si además en la anamnesis se recogen ronquidos, apneas nocturnas, sudación nocturna excesiva, hiperextensión del cuello durante el sueño o somnolencia diurna, está indicada la remisión al segundo nivel. El tratamiento con presión positiva continua de la vía aérea, que requiere un esfuerzo de adaptación por parte del paciente y de su familia, y el tratamiento quirúrgico son las opciones más habituales21. La adenoamigdalectomía puede ser una técnica insuficiente por la estrechez generalizada de la vía aérea de los pacientes con SD, por lo que en ocasiones se ofertan intervenciones más ampliadas como la uvulopalatofaringoplastia.
La enfermedad periodontal es característica en estos pacientes y aumenta con la edad. Depende de un conjunto de factores como la mala higiene bucal, las malposiciones dentarias, la macroglosia relativa y el bruxismo. Las visitas periódicas al dentista y la higiene bucal favorecen la conservación de piezas dentarias.
Alteraciones oftalmológicas
La patología oftalmológica en el SD se puede manifestar desde el momento del nacimiento. Antes de los 6 meses de edad debería realizarse una primera revisión para descartar la presencia de cataratas congénitas y glaucoma. Posteriormente aparecen los defectos de refracción (35-76%), el estrabismo (25-57%) y el nistagmo (20%)22. Como todos estos procesos pueden ir manifestándose gradualmente, se aconseja el seguimiento por parte del oftalmólogo con una periodicidad inicialmente anual y posteriormente, si no se detectan anomalías, de forma bienal. A partir de los 20 años aparecen otras patologías como las cataratas o el queratocono. El médico de familia debe permanecer atento vigilando periódicamente la agudeza visual o remitiendo al paciente al oftalmólogo para que no pasen desapercibidas.
Trastornos ortopédicos
La inestabilidad atlantoaxoidea es una entidad casi siempre asintomática que puede diagnosticarse en hasta el 20% de los pacientes con SD. Depende de una laxitud ligamentosa, característica en este síndrome, que afecta en este caso a los ligamentos que fijan las dos primeras vértebras cervicales. Se demuestra practicando un estudio radiológico de la columna cervical de perfil, en posición neutra, en flexión y en hiperextensión, objetivando una distancia entre la cara posterior del axis y la anterior de la odontoides superior a 5 mm en cualquiera de las tres radiografías. Habitualmente el espacio atlanto-axoideo aumenta con la flexión cervical. En un pequeño porcentaje de pacientes portadores de esta anomalía (1-2%) se desarrolla una compresión medular sintomática que se manifiesta con dolor cervical, trastornos del equilibrio, pérdida de fuerza en extremidades o alteraciones esfinterianas. En determinadas situaciones de estrés, que incluyen traumatismos deportivos e intubación endotraqueal, la clínica puede manifestarse de forma dramática poniendo incluso en peligro la vida del paciente. Esta alteración saltó a un primer plano en 1983 cuando el Medical Advisory Committee of the Special Olympics exigió a los participantes en determinados deportes de riesgo el cribado radiológico para autorizar la inscripción. Posteriormente se ha postulado el cribado universal en personas con SD, aunque hay partidarios de vigilar periódicamente en estos pacientes los signos de compresión medular y emprender sólo los estudios diagnósticos ante situaciones de riesgo, fundamentalmente quirúrgico.
Otros problemas ortopédicos han recibido menor atención aunque probablemente supongan un volumen mucho mayor de consultas para el médico de familia. La laxitud ligamentosa y la hipotonía favorecen la aparición de esguinces, y los pies planos y el genu valgo son muy frecuentes.
Demencia
La mayoría de personas con SD desarrollarán una demencia tipo Alzheimer si viven lo suficiente. A diferencia de lo que sucede en la población general, el déficit cognitivo se instaura a una edad temprana. Coppus et al., en un estudio de base poblacional, encontraron una prevalencia global del 16,8% entre los individuos de más de 45 años, con un pico del 32% entre los 50 y los 59 años23. La clínica de comienzo se caracteriza por alteraciones del comportamiento y de la personalidad, y en ocasiones aparece un síndrome disejecutivo frontal que se manifiesta por dificultad o desinterés en la realización de tareas habituales24. Estos cambios pueden ser atribuidos erróneamente a sintomatología depresiva, por lo que la ausencia de respuesta a una dosis adecuada de un inhibidor selectivo de la recaptación de serotonina puede ayudar a enfocar al paciente. Las alteraciones de la memoria suelen aparecer en un segundo plano. El sobrediagnóstico debe evitarse cuidadosamente y los pacientes con SD sospechosos de padecer déficit cognitivo deben ser remitidos para su estudio al segundo nivel.
Actividades preventivas y promoción de la salud
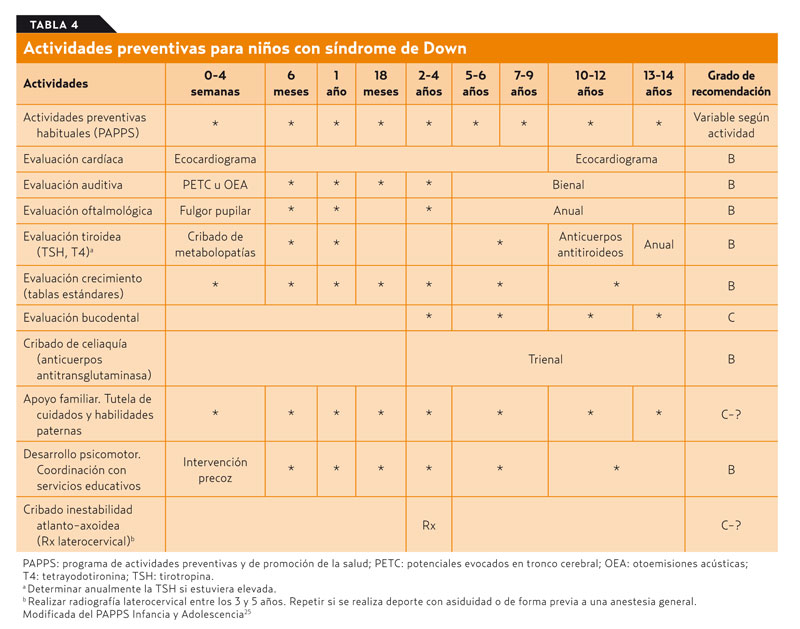
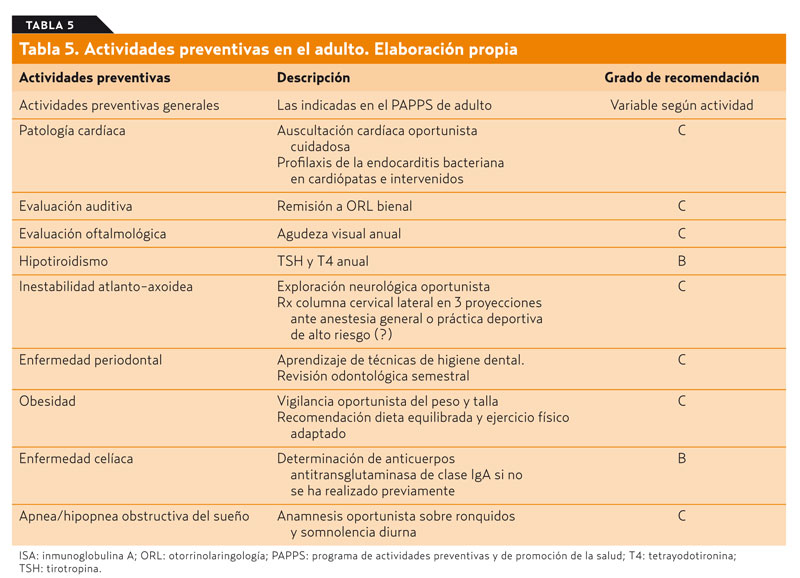
Las personas con SD deberían tener los mismos cuidados preventivos que la población general. El grupo PAPPS de la Infancia y Adolescencia recomienda una serie de medidas específicas que se relacionan en la tabla 425. Estas actividades tienen su continuidad en la vida adulta. En la tabla 5 se detallan una serie de actividades preventivas que parece razonable aplicar a este grupo de población, aunque la gran mayoría de ellas sólo cuentan con un grado de recomendación C.
La promoción de la salud tampoco debería diferir sustancialmente de la diseñada para el conjunto de la población. Resulta útil aprovechar las visitas a la consulta para incidir en aspectos relacionados con sus necesidades. Una dieta equilibrada y la práctica regular de ejercicio físico ayudan a contrarrestar su tendencia a la obesidad. Resulta muy conveniente supervisar e impulsar la cumplimentación de una adecuada higiene bucodental. Igualmente conviene explorar, en sintonía con los padres, las necesidades sexuales expresadas para poder ofertar las respuestas que se consideren en cada caso.
¿Qué hacemos con Juan?
El oftalmólogo comunica que se ha incluido a Juan en la lista de espera quirúrgica para ser intervenido de cataratas. Los resultados analíticos en los que se solicitaron hormonas tiroideas y anticuerpos antitransglutaminasa son normales. Se aprovecha para interrogar al paciente y a su familia sobre dificultades en la audición, presencia de ronquidos y apneas durante el sueño y somnolencia diurna. Se practica una exploración general que incluye pesado, otoscopia, auscultación cardíaca cuidadosa, exploración neurológica centrada en reflejos osteotendinosos en extremidades y examen de la boca. La anamnesis y la exploración física resultan normales, únicamente se objetiva malposición dentaria y aspecto enrojecido y tumefacto del borde gingival. Se remite a Juan a la consulta de enfermería para ser instruido en técnicas de higiene bucodental y, de acuerdo con la familia, se decide realizar una visita al odontólogo.
Lecturas recomendadas
Soriano Faura FJ. Actividades preventivas en niños con síndrome de Down. En: Recomendaciones PrevInfad / PAPPS [en línea]. Actualizado abril de 2007 [consultado el 11/10/2010]. Disponible en: http://www.aepap.org/previnfad/Down.htm
Excelente revisión de la indicación de actividades preventivas en niños con síndrome de Down con un enfoque basado en la evidencia.
Smith DS. Health care management of adults with Down syndrome. Am Fam Physician.2001;64(6):1031-9.
Artículo de revisión que describe el manejo de los pacientes adultos con síndrome de Down desde la perspectiva del médico de familia.
Borrel JM, Florez J, Seréz A, Fernández R, Albert J, Prieto C, et al. Programa español de salud para personas con síndrome de Down [en línea]. Actualizado en 2010. [consultado el 11/10/2010]. Disponible en: http://www.sindromedown.net/adjuntos/cPublicaciones/64L_programa.pdf
Instrumento de apoyo a las familias y al personal sanitario que repasa los aspectos fundamentales de la problemática del síndrome de Down de un modo práctico y ameno.
Bibliografía
- Down JLH. Observations on an ethnic classification of idiots. London Hospital Reports. 1886;3:259-62.
- Cuckle HS, Wald NJ, Thompson SG. Estimating a women´s risk of having a pregnancy associated with Down´s syndrome using her age and serum alpha-protein level. Br J Obstet Gynaecol. 1987;94:387-402.
- Bermejo E, Cuevas L, Mendioroz J, Grupo Periférico del ECEMC y Martínez-Frías ML. Frecuencia de anomalías congénitas en España: Vigilancia epidemiológica en el ECEMC en el período 1980-2008. Rev Dismorfología Epidemiología. 2009;(8):64-91.
- Morris JK, Alberman E. Trends in Down´s syndrome live births and antenatal diagnoses in England and Wales from 1989 to 2008: analysis of data from the National Down Syndrome Cytogenic Register. BMJ. 2009;b3794. doi:10.1136/bmj.b3794
- Leonard S, Bower C, Petterson B, Leonard H. Survival of infants born with Down´s syndrome: 1980-96. Paediatr Perinat Epidemol. 2000;14(2):163-71.
- Bittles AH, Glasson EJ. Clinical, social and ethical implications of changing life expectancy in Down syndrome. Dev Med Child Neurol. 2004;46(4):282-6.
- Wilson RD, Langlois S, Johnson JA; Society of Obstetricians and Gynaecologists of Canada. Mid trimester amniocentesis fetal loss rate. J Obstet Gynaecol Can. 2007;29:586-95.
- Nicolaides KH, Spencer K, Avgidou K, Faiola S, Falcon O. Multicenter study of first-trimester screening for trisomy 21 in 75821 pregnancies: results and estimation of the potential impact of individual risk-orientated two stage first-trimester screening. Ultrasound Obstet Gynecol. 2005;25:221-6.
- Spencer K, Souter V, Tul N, Snijders R, Nicolaides KH. A screening program for trisomy 21 at 10-14 week using fetal nuchal translucency, maternal serum free beta-human chorionic gonadotropin and pregnancy-associated plasma protein-A. Ultrasound Obstet Gynecol. 1999;13:231-7.
- Spencer K, Spencer CE, Power M, Dawson C, Nicolaides KH. Screening for chromosomal abnormalities in the first trimester using ultrasound and maternal serum biochemistry in a one-stop clinic: a review of three years prospective experience. Br J Obstet Gynaecol. 2003;110:281-6.
- Tolmie JL. Down syndrome and other autosomal trisomies. In: Principles and Practice of Medical Genetic. 3 ed. New York: Churchill Livingstone; 1996. p. 925-72.
- National Down Syndrome Society. Hitos del desarrollo [consultado el 21/11/2010]. Disponible en: http://esp.ndss.org/images/stories/NDSSresources/pdfs/hitos_de_desarrollo.pdf
- Pelchat D, Bisson J, Ricard N, Perreault M, Bouchard JM. Longitudinal effects of an early family intervention programme on the adaptation of children with a disability. Int J Nurs Stud. 1999;36:465-77.
- Pastor X, Quintó L, Corretger M, Gassió R, Hernández M, Serés A. Tablas de crecimiento actualizadas de los niños españoles con síndrome de Down. Revista Médica Internacional sobre el Síndrome de Down. 2004;8(3): 34-46.
- Karlsson B, Gustafsson J, Hedoy G, Ivarsson S-A, Annerén G. Thyroid dysfunction in Down´s syndrome: relation to age and thyroid autoimmunity. Arch Dis Child. 1998;79:242-5.
- Swigonski NL, Kuhlenschmidt HL, Bull MJ, Corkins MR, Downs SM. Screening for Down syndrome: cost-effectiveness of preventing lymphoma. Pediatrics 2006;118:594-602.
- Vis JC, de Bruin-Bon RH, Bouma BJ, Huisman SA, Imschot L, van den Brink K, Mulder BJ. Congenital heart defects are under-recognised in adult patients with Down´s syndrome. Heart. 2010;96:1480-4.
- Goldhaber SZ, Brown WD, Sutton MG. High frequency of mitral valve prolapse and aortic regurgitation among asymptomatic adults with Down’s syndrome. JAMA. 1987;2;258:1793-5.
- Venail F, Gardiner Q, Mondain M. ENT and Speech Disorders in Children with Down’s Syndrome: an Overview of Pathophysiology, Clinical Features, Treatments, and Current Management. Clin Pediatr. 2004;43(9):783-91.
- Shott SR, Amin R, Chini B, Heubi C, Hotze S, Akers R. Obstructive sleep apnea. Should all children with Down syndrome be tested?. Arch Otolaryngol Head Neck Surg. 2006;132:432-6.
- de Miguel-Díez J, Villa-Asensi JR, Álvarez-Sala JL. Prevalence of sleep-disordered breathing in children with Down syndrome: polygraphic findings in 108 children. Sleep. 2003;26:1006-9.
- Roizen NJ, Patterson D. Down’s syndrome. The Lancet. 2003;361:1281-9.
- Coppus A, Evenhuis H, Verberne GJ, Visser F, van Gool P, Eikelenboom P, van Duijin C. Dementia and mortality in persons with Down´s syndrome. J Intellect Disabil Res. 2006;50:768-77.
- Ball SL, Holland AJ, Hon J, Huppert FA, Treppner P, Watson PC. Personality and behavior changes mark the early disease in adults with Down´s syndrome: findings from a prospective population-based study. Int J Geriatr Psychiatry. 2006;21:661-73.
- Soriano Faura FJ. Actividades preventivas en niños con síndrome de Down. En Recomendaciones PrevInfad/PAPPS [en línea]. Actualizado abril de 2007 [consultado el 11/10/2010]. Disponible en: http://www.aepap.org/previnfad/Down.htm
Ojuel 02-04-11
Muy interesante. Añado este enlace que habla sobre la sexualidad y la afectividad, tanto en la adolescencia como en la edad adulta de las personas con síndrome de down, que es un tema que se suele evitar por la 'infantilización' que se les ha supuesto. Forma parte de un blog sobre sexualidad en situaciones 'especiales' que es interesante revisar.http://sexualidadespecial.blogspot.com/2010/09/edad-adulta-sexualidad_23.htmljulia ojuel solsona, médica de família, badalona